Facility Equipment
Pharmacists have been providing sterile compounding services in institutions for decades. These services have provided parenteral therapies, infusion services, and complex infusion administration devices and supplies. However, in the past two decades, compounding sterile formulations and providing administration services has expanded beyond the institution. These additional areas include home care agencies, infusion service agencies, outpatient clinics, and community pharmacies. Pharmacists are also providing patient and caregiver assessments, education, and skills, and are taking the responsibility for coordinating patient care through an interdisciplinary team.
Pharmacists will compound a wide variety of sterile formulations in these different settings. These formulations will include products administered by injection (IV, IM, SQ, ID, intrathecal, epidural) or via inhalation, intranasal, or ophthalmic routes of administration. Sterile formulations for either institutional or home care use have a number of special requirements such as:
- sterility
- particulate material
- pyrogen free
- stability
- pH
- osmotic pressure
Sterility is the freedom from bacteria and other microorganisms. Formulations must be sterile, which is not a relative term; an item is either sterile or not sterile. It is not possible to have a compounded preparation (or manufactured product) to be 100% sterile or to check every manufactured or compounded dosage form for sterility since the results only apply to the actual samples tested. According to USP-NF General Chapter <71> Sterility Tests, there are several requirements for conducting sterility testing:
- cleaning and disinfection of the aseptic work area
- air quality and surface testing for microbial contamination
- glove imprint testing
- visual confirmation that personnel are properly donning protective garments
- certification of personnel knowledge and compounding dexterity
There are two official sterility testing methods. The minimum number of samples to be tested in relation to the number of CSP preparations compounded in a batch is given in Table 3 in General Chapter <71>. The number of samples to be tested depends on the size and type of CSP and the number of those items in the batch.
The first one filters a solution through a 0.45 micron filter to collect microbes on the filter. Then all or part of the filter is placed into containers with suitable media. The second method is a direct inoculation of the CSP sample on to the culture media.
The test results are based on the turbidity of the culture media. After 14 days, if the media remains clear, this indicates that there is no bacteria growth thus the preparation passes the sterility test. But if the media indicates microbial contamination, the test can be repeated using the same number of samples as the original testing. If the repeat test shows no growth, the preparation is judged to pass the sterility test.
General Chapter <797> Pharmaceutical Compounding-Sterile Preparations states that sterility testing outlined in Chapter <71> may be applied to low-risk level and medium-risk level CSPs. But for high-risk level preparations, a standard self-contained biological indicator should be added to non—dispensable specimens before terminal sterilization to determine whether the sterilization cycle was adequate.
If the sterile formulation is a solution, it must be free of all visible particulate material. Particulate materials refer to the mobile, undissolved substances unintentionally present in parenteral products. Examples of such material are cellulose, glass, rubber cores from vials, cloth or cotton fibers, metal, plastic, rust, diatoms, and dandruff.
It is not possible to have preparations 100% free of particulate matter, so General Chapter <788> Particulate Matter in Injections has defined two methods to count the number of particulates in injections. The light obscuration particle count method operates on the principle of light blockage and is calibrated with particles of 10 microns and 25 microns. The microscopic particle count method uses a microscope with an ocular micrometer at a magnification factor of 100.
Sterile suspensions and ointments may have particulate material, but these are usually the active drug or an ingredient, not contaminants. There, these dosage forms are generally exempt from the requirements of the chapter.
Particles measuring 50 microns or larger can be detected by visual inspection. Specialized equipment is needed to detect particles less than 50 microns in size. The USP 24/NF19 Section <788> sets limits on the number and size of particulates that are permissible in parenteral formulations. For large volume parenterals, the limit is not more than 12 particles/ml that are equal to or larger than 10 microns, and not more than 2 particles/ml that are equal to or larger than 25 microns. For small volume parenterals, the limit is 3000 particles/container that are equal to or larger than 10 microns, and not more than 300 particles/container that are equal to or larger than 25 microns.
The potential sources of particular material are:
- The product itself
- Manufacturing and such variables as the environment, equipment, and personnel
- The packaging components
- The administration sets and devices used to administer the product
- The manipulations and environment of the product at the time of administration.
Sterile formulations must be pyrogen free. Pyrogens are metabolic by-products of living microorganisms. So if pyrogens are detected in a sterile product, that means that bacteria have proliferated somewhere along during the formulation process. In humans, pyrogens cause significant discomfort but are rarely fatal. Symptoms include fever and chills, cutaneous vasoconstriction, increased arterial blood pressure, increased heart workload, pupillary dilation, piloerection, decreased respiration, nausea and malaise, severe diarrhea, or pain in the back and legs.
The stability of drugs in sterile formulations is an important consideration. In institutional settings, most admixtures are prepared hours in advance of when they are to be administered, and are generally utilized within a short period of time. In home health care settings, admixtures are prepared days in advance of when they are to be administered, and are generally utilized over longer periods of time compared to the clinical setting. Therefore, the stability of a particular drug in a particular sterile formulation must be known.
Physiological pH is about 7.4, and an effort should be made to provide sterile formulations that do not vary significantly from that normal pH. Of course, there are situations in which this becomes a secondary consideration because acidic or alkaline solutions may be needed to solubilize drugs or used as a therapeutic treatment themselves.
Osmotic pressure is a characteristic of any solution that results from the number of dissolved particles in the solution. Blood has an osmolarity of approximately 300 milliosmoles per litter (mOsmol/L), and ideally any sterile solution would be formulated to have the same osmolarity. The most commonly used large volume parenteral solutions have osmolarities similar to that of blood; for example, 0.9% sodium chloride solution (308 mOsmol/L) and 5% dextrose solution (252 mOsmol/L).
Intravenous solutions that have larger osmolarity values (hypertonic) or smaller osmolarity values (hypotonic) may cause damage to red blood cells, pain, and tissue irritation. However, there are some therapeutic situations where it may be necessary to administer hypertonic or hypotonic solutions. In these cases, the solutions are usually given slowly through large veins to minimize the reactions.
A critical site is any location that is exposed and can allow intrusion of microorganisms and/or foreign matter. General Chapter <797> defines a critical site as, “A location that includes any component or fluid pathway surfaces (e.g., vial septa, injection ports, beakers) or openings (e.g., opened ampules, needle hubs) exposed and at risk of direct contact with air (e.g., ambient room or HEPA filtered), moisture (e.g., oral and mucosal secretions), or touch contamination.”
Primary Engineering Control (PEC)
Chapter <797> defines Primary Engineering Control (PEC) as a device or room that provides an ISO Class 5 environment for the exposure of critical sites when compounding CSPs. Such devices include, but may not be limited to, laminar airflow workbenches (LAFWs), biological safety cabinets (BSCs), compounding aseptic isolators (CAIs), and compounding aseptic containment isolators (CACIs).
| ISO Class | Particle Count (per m3)* |
| 3 | 35.2 |
| 4 | 352 |
| 5 | 3,520 |
| 6 | 35,200 |
| 7 | 352,000 |
| 8 | 3,520,000 |
| *The particles are 0.5 micron or larger. | (3,520 particles per m3 is equivalent to 100 particles per ft3) |
If the PEC is a clean room, there are two general approaches and the method depends on the layout of the clean room facilities. One clean room design has a separate room for the buffer area and the ante-area with a barrier between them. The second design has the buffer area and ante-area combined. In this second design, the buffer area receives the “first air,” and that air washes over the ante-area before exiting. This design is called the “displacement approach” and cannot be used for high-risk level compounding. When used for low- and medium-risk level compounding, the proper air flow must be maintained from the cleaner air downstream, with no turbulent or stagnant areas.
| Risk Level | Equipment | Examples |
|---|---|---|
| Low Risk | ISO Class 5 LAFW in ISO Class 8 clean room with anteroom |
|
| Medium Risk | ISO Class 5 LAFW in ISO Class 8 clean room with anteroom |
|
| High Risk | ISO Class 5 LAFW in Class 8 clean room with a separate anteroom |
|
One literal application of Chapter <797> definitions would be performing all sterile compounding inside an ISO Class 5 device. The chapter lists four such devices: laminar airflow workbenches (LAFWs), biological safety cabinets (BSCs), compounding aseptic isolators (CAIs), and compounding aseptic containment isolators (CACIs).
Laminar Air Flow Workbenches (Horizontal and Vertical LAFW)
Laminar flow hoods are used to control airborne contamination of sterile products during their extemporaneous preparation. The direction of air flow may be horizontal or vertical. Horizontal flow hoods are most commonly used, with the more costly vertical flow hoods being reserved for agents that may produce an environmental hazard (e.g. cytotoxic agents, radioactive agents, antimicrobial agents).
 |
 |
|
|
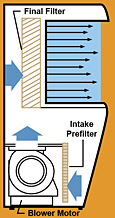
Horizontal Flow Hood
|
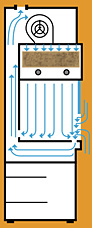
Vertical Flow Hood
|
In horizontal LAFW, room air is drawn into the hood through a prefilter to remove relatively large contaminates such as dust and lint. Then the air is filtered through a high efficiency particulate air (HEPA) filter removing 99.97% of all particles 0.3 microns or larger. Parallel air streams bathe the work area with a velocity of 80-100 ft/min which is sufficient to provide the area free of particles and microorganisms and prevent room air from entering the work area.
In vertical laminar airflow workstations, the filtered air enters at the top of the work area and moves downward. In some models the air moves downward all the way through the work area before it is returned to the room air. In other models the air moves downward initially but then turns inside the work area and exits from the hood through the opening at the front of the hood.
Laminar airflow workbenches used in sterile compounding must be ISO Class 5. They are effective only when properly used. Interruption of the air flow will interfere with the effectiveness of the hood. Downstream contamination occurs when any object comes between the HEPA filter and the sterile product, interrupting the parallel flow and creating dead space. Cross-stream contamination may occur due to rapid movements of the operator in the hood. Backward contamination may be caused by turbulence created by objects being placed in the hood, by fast traffic passing the hood, or by coughing, sneezing, etc. by the operator.
|
Dead space created around object in laminar flow hood |

It should be remembered that the hood does not produce sterilization, but merely prevents contaminants from settling onto the surface of the sterile product. Any movement of greater velocity and different direction than that of the hood’s air flow will create a turbulence that reduces the hood’s effectiveness. Contamination may be minimized by working at a smooth, steady pace at least 6 inches into the hood.
Maintaining laminar airflow workstations is essential. Changing prefilters and HEPA filters, routine cleaning of the hood, and any other maintenance should be done according to the manufacturer’s recommendations and time schedules. A quality control standard operating procedure (SOP) should be developed and followed to ensure that maintenance is done when required and to document that maintenance has indeed been done. These steps should be documented.
Biological Safety Cabinets
LAFW blows air toward operator whereas biological safety cabinets (BSCs) provide protection for both the personnel and the environment. They are designed to take air into the cabinet through a prefilter and channel it through a HEPA filter located in the top of the cabinet. The airflow is directed downward toward the work surface like the vertical LAF. As the air approaches the work surface, it is pulled through vents at the front, back, and sides of the cabinet. The air is channeled so a major portion is recirculated back into the cabinet and a minor portion is passed through another HEPA filter before being exhausted into the room.
Due to its advantages over LAFW (i.e. protection for both the personnel and the environment), BSCs should be used when preparing chemotherapy and hazardous drugs. There are a number of different types of BSCs listed in the table below. All classes have HEPA/ULPA filter.

Maintaining BSCs is essential but similar to what was described in the LAFW section.
Isolators
General Chapter <797> defines two types of isolators and requires any isolator used for sterile compounding to maintain an ISO Class 5 environment:
- A Compounding Aseptic Isolator (CAI) is designed to maintain an aseptic compounding environment for ingredients within the isolator throughout the compounding and material transfer processes. Air exchange into the isolator from the surrounding environment should not occur unless the air has first passed through a microbial retentive filter (HEPA minimum).
- A Compounding Aseptic Containment Isolator (CACI) is designed to protect workers from exposure to undesirable levels of airborne drug throughout the compounding and material transfer processes and to provide an aseptic environment for compounding sterile preparations. Where volatile hazardous drugs are prepared, the exhaust air from the isolator should be appropriately removed by properly designed building ventilation.
Compounding isolators take advantage of an airtight glove/glove port design that allows the user to perform hands-on tasks inside the isolator without compromising the intended performance of the isolator. There are many variables in the design of isolators because there are no uniform industry standards for its manufacture. But they usually follow the following objectives:
- the full enclosure of the drug compounding process
- the intentional use of air pressure to define the direction of airflow in/out of the cabinet
- the use of airflow capture velocities to capture and remove aerosolized drug product near its point of generation
- the use of HEPA to capture aerosolized drug preparations and particulate contamination
- the use of external venting to remove vaporized hazardous drugs from the work chamber and from the pharmacy
- the use and material transfer processes that allow material transfer in/out of the compounding isolator without compromising worker exposure to undesirable levels of airborne drug or unwittingly compromising the sterility of the compounding environment.
Clean Rooms
One method of protecting the critical sites is to use a device that creates an ISO Class 5 environment, a direct compounding area (DCA), and completing all of the compounding operations inside that device. LAFWs, CAIs, and CACIs are examples of DCA. A cleanroom is another example because the entire room is the DCA with ISO Class 5 environment.
If the cleanroom is an ISO Class 5 environment, there need to be additional barriers around the room that reduce the particle burden, and provide a staging area for the materials and activities necessary to prepare CSPs. The additional barrier is called a buffer area. Air quality in this area is ISO Class 7 and the activities appropriate for the area include the preparation and staging of components and supplies. The ante-area is the area outside of buffer area and should have ISO Class 7 or 8 air quality. Activities in this area include personnel hand hygiene and garbing procedures, staging of components, order entry, CSP labeling and other high particulate generating activities.
The proper design of cleanrooms must take in to consideration work flow, efficiency and personnel comfort factors such as temperature, lighting and noise. The risk level of the CPS also is a consideration. What’s more, Chapter <797> allows several exceptions to installing a cleanroom. For example, immediate-use CSPs can be prepared at the patient’s bedside or in fast-paced treatment areas such as emergency rooms, operating rooms, therapeutic radiology, cardiac catherization, and respiratory therapy. Allergan extracts may be prepared in allergy clinics as long as standards are met. Pharmacy satellites and outpatient treatment centers preparing low-risk level CSPs with 12-hour or less beyond-use-dates are exempt as well. These CSPs can be compounded in segregated areas without an ante-area. However, the segregated area must contain a device that provides unidirectional airflow of ISO Class 5 quality air. The area should also limit personnel activities and traffic, and avoid materials that are extraneous to sterile. The pharmacy in those settings must place ISO Class 5 PEC in a restricted access buffer area with an ISO Class 7 environment if low-, medium-, or high-risk level aseptic compounding is to be performed. Last but not least, pre-sterilization procedures for high-risk level CSPs must be completed in no worse than an ISO Class 8 quality air environment.